依据欧盟MDR法规要求,所有医疗器械(除少数豁免外)均需通过临床评价来证明其符合安全与性能要求。对于新手而言,临床评价计划、文献检索、临床评价报告的先后顺序是什么呢?本文将基于实操经验,系统性梳理CE临床评价的标准操作流程。
第一步:制定临床评价计划(Clinical Evaluation Plan, CEP)
临床评价的起点并非数据收集,而是计划制定。MDR要求制造商必须建立并持续更新临床评价计划。CEP应明确界定评价的范围、预期用途、目标人群、临床参数(安全性和性能指标)、数据来源策略以及评价方法。
在这一阶段,制造商即应开始考虑等同性策略——如果计划采用等同性路径,就需要在市场调研阶段锁定潜在的等同器械,并评估其临床数据的可获取性。
第二步:识别相关临床数据(Identification of Pertinent Data)
数据识别是临床评价的第一阶段,可用于临床评价和等同性论证的数据来源包括:
(1)申请器械自身的临床试验数据;(2)等同器械的临床试验或在科学文献中报道的研究数据;(3)发表在同行评议科学文献上的、关于申请器械或等同器械的临床经验报告;(4)通过上市后临床随访(PMCF)活动产生的临床数据。
第三步:评价数据的科学有效性(Appraisal of Pertinent Data)
识别出相关数据后,需要对其方法学质量、科学有效性和临床相关性进行系统评价。这一阶段的核心是区分哪些数据可以作为等同性论证的可靠依据,哪些数据因方法学缺陷而应予排除。
第四步:等同性论证——核心环节
这是整个临床评价中技术含量最高、也最容易被公告机构质疑的环节。
1、筛选等同器械
制造商需要从市场上已获CE认证的器械中筛选潜在的等同器械。建议选择市场声誉良好、文献数据充分的器械作为对标对象。选择时不仅要关注技术上的可比性,还要关注与等同器械相关的科学文献的数量和质量。
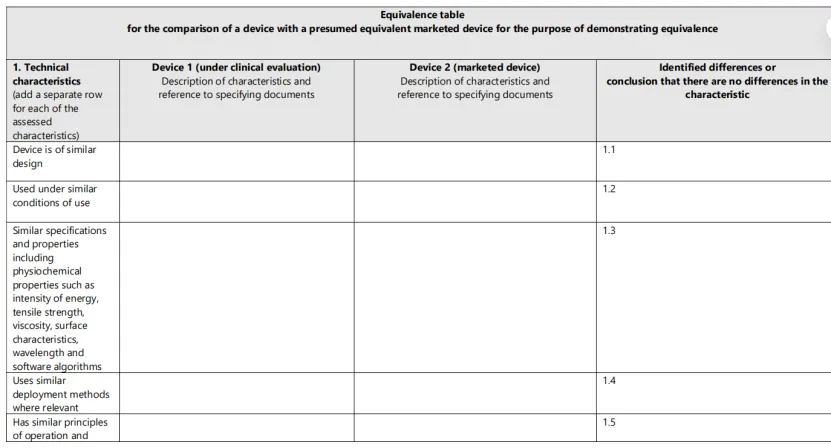
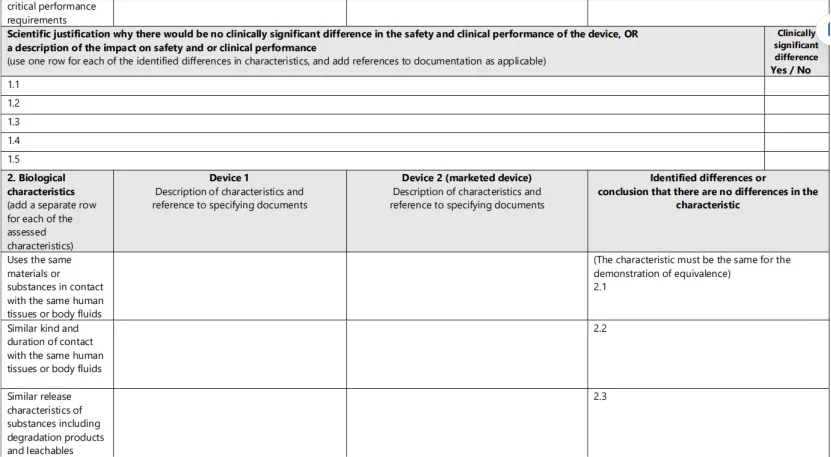
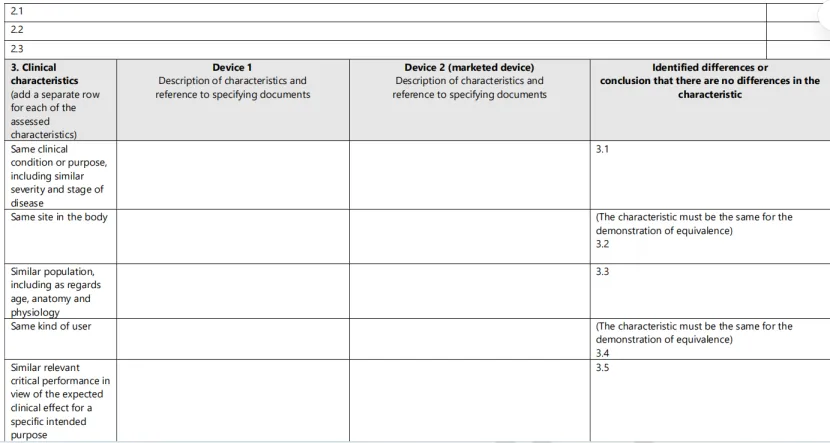
2、填写等同性对比表
MDR要求制造商逐项对比技术、生物、临床三个维度的所有特性。这是公告机构审查的核心文件。详见附录:临床评价等同性对比表模板。
3、差异分析与科学论证
MDCG 2020-5明确指出,等同性的本质是“相似”而非“相同”,但任何差异都必须基于科学论证,证明其不会导致临床安全性和性能的显著差异。
对于有差异的项目,制造商可以通过额外的测试、动物实验、科学文献检索等方式来证明差异对临床安全和性能的影响。某些差异,例如人工智能产品的核心算法差异,可能需要通过临床研究数据来证明。
4、数据访问权的保障
如果申请器械与等同器械属于不同制造商,且申请器械为III类或植入式器械,则MDR Article 61(5)要求制造商必须与等同器械的制造商签订技术文档共享合同,确保可持续访问等同器械的完整技术文件。这一要求在实际操作时有重大挑战——2023年曾有德国心脏起搏器制造商因合同中未明确“持续访问权”而被公告机构拒绝等同性认定。
第五步:编写临床评价报告(CER)
在完成数据识别、评价和等同性论证后,制造商需将所有分析整合入CER。
CER不是简单的数据罗列,而是一份系统的科学论证文件。它需要清晰地说明:申请器械是什么、预期用途是什么、有哪些临床数据可用、这些数据如何证明器械的安全性和性能、风险-收益平衡是否可接受。如果采用等同性路径,CER还需详细论证等同性成立的依据,并明确列出被引用数据的来源和访问方式。
第六步:上市后临床随访(PMCF)--临床评价的延续
MDR的一个重要变革是将PMCF定义为临床评价的持续更新过程。制造商必须主动收集和评估已上市器械的临床数据,以确认器械在整个预期使用寿命内的安全性和性能。
对于通过等同性论证获批的器械而言,PMCF具有特殊的重要性。由于申请器械自身的直接临床数据有限,PMCF成为验证等同性结论、发现潜在差异、积累自身临床证据的关键手段。
我们有丰富的医疗器械国内外认证经验,更多医疗器械国内注册、CE认证、FDA认证、培训等信息,请添加微信:cgrz6199 欢迎进一步沟通!
附录:临床评价等同性对比表模板